
期刊:Science China-Life Sciences
影响因子:8.0
单细胞测序概述
单细胞测序(Single-cell RNA sequencing; scRNA-seq)是一种开创性的单细胞测序技术,现在已得到广泛普及,并涵盖了多种方法。尽管方法多种多样,但它们都遵循类似的一般过程,包括四个主要步骤:(i)单细胞分离、(ii)逆转录(RT)、(iii)cDNA 扩增和(iv)测序文库构建和测序(Hedlund and Deng,2018)。scRNA-seq工作流程的主要步骤如图 3所示。本节概述了与单细胞分离和测序文库构建相关的一些技术和解决方案。
(1)单细胞分离
在 scRNA-seq中,第一步也是关键的一步是单细胞分离,其中组织解离和单细胞分离被认为是造成污染、批次效应和程序差异的重要因素(Tung等,2017)。因此,要进行高通量和无偏差的单细胞测序,高效、可靠、准确捕获单细胞是关键决定因素。早期的单细胞分离方法包括有限连续稀释 ( Gross等,2015 )、手动显微操作 ( Hu等,2016a )和激光捕获显微切割 (LCM)( Emmert-Buck等,1996 ),这些方法通量低、耗时、效率低、技术难度大,但仍用于分析少量细胞(例如稀有细胞)( Dal Molin and Di Camillo, 2019 )。
流式细胞荧光分选 (FACS)是一种常用的高通量技术,可特异性地自动分离数千个单个细胞,但对细胞数量的需求很大(需要分离的细胞数量 >10,000)( Hu等,2016a )。此外,由于荧光信号微弱,该技术不适用于某些标志物表达较低的细胞,因此很难区分具有相似标志物表达的亚群 ( Yasen等,2020 )。磁激活细胞分选 (MACS)是另一种高通量分离技术,旨在通过与磁珠结合的酶、凝集素、抗体或链霉亲和素分离各种细胞类型,从而促进特定蛋白质与靶细胞的结合 ( Hu等,2016a )。MACS 系统具有显著的优势,可在特定细胞群中实现 90% 以上的纯度 ( Miltenyi等,1990 )。然而,与 FACS 相比,MACS 具有固有的局限性,因为免疫磁技术只能将细胞分离为阴性和阳性细胞群。此外,它不能根据分子的低表达或高表达来分离细胞,而 FACS 可以做到这一点 ( Hu等,2016a )。在当前的高通量测序平台中,基于微流体的单细胞操作方法已成为转录组研究中单细胞分离的主要技术,这种技术显著提高了分离过程的规模、效率和准确性。微流体装置使用反应室或液滴捕获细胞,然后分别进行单独的纳升水平的反应步骤,提供了更经济且样品效率更高的分析方法。这些装置主要分为基于微孔的方法、基于液滴的方法和集成流体回路 (IFC)。微流控系统在 scRNA 测序中的集成显著提高了测序通量,能够同时处理和分析数以万计的单细胞。表S2 全面概述了当前的单细胞分离技术,包括其优点和局限性。
(2)逆转录和 cDNA 扩增
单个哺乳动物细胞含有的 RNA总量约 10 pg,其中主要部分由核糖体RNA (rRNA)和转运RNA (tRNA)组成,而信使RNA (mRNA)仅占总量的1%–5%(Liu等,2014 ; Wang等,2023c)。由于单细胞中mRNA含量极低,因此必须在逆转录后对cDNA进行扩增,以获得大量用于测序文库制备的样本。cDNA扩增可以通过使用聚合酶链式反应 (PCR)的指数扩增或基于体外转录 (IVT)的线性扩增来实现。
目前,文库构建的主要方法是基于 PCR 的 cDNA 扩增,包括 poly(A)加尾和模板转换 (TS)方法(Kolodziejczyk等,2015)。poly(A)加尾法使用 oligo-dT 引物与 mRNA 3′-poly(A)尾结合,将mRNA 逆转录为 cDNA。poly(A)加尾法速度快但不能捕获非多聚腺苷酸化(Poly(A)−)RNA(Hebenstreit,2012),且捕获效率低,文献报道现行捕获方法的捕获效率约为10%–15%(Islam等,2014)。此外,逆转录酶反应的终止还可能导致转录中 mRNA 5′端的覆盖率降低。TS 技术利用莫洛尼鼠白血病病毒 (MMLV)逆转录酶进行 TS 过程。MMLV 逆转录酶可以在新合成的单链尾端附加一个 poly(C)尾。poly(C)尾可以与模板转换寡核苷酸 (TSO)接头序列的 5′端 poly(G)尾结合。在这一互作之后会发生一个“切换”:逆转录酶以TSO为模板合成cDNA,完成衔接子的转换(Picelli,2017)。TS法相对来说不易发生核酸丢失,但与poly(A)加尾法相比灵敏度较低。基于PCR的扩增虽然能够在短时间内快速有效地扩增大量cDNA,但受到指数扩增过程固有特性的制约。PCR倾向于扩增较短和GC含量较低的扩增子,从而导致定量偏差和非特异性转录本的积累,从而导致原始转录本信息的丢失(Aird等,2011)。此外,PCR会导致最终文库中高表达的转录本过多(Aird等,2011)。
IVT代表了另一种cDNA合成和扩增的方法。IVT 的主要优势在于其线性扩增,这一特性可减轻人们通常所说的扩增偏好,使其比 PCR 更精确、重复性更好 ( Chen等,2017a ; Grün and van Oudenaarden, 2015 )。在该策略中,一条oligo-dT 引物最初与 mRNA 的 3′-poly(A)尾结合,合成两条 cDNA 链,该引物包含(i)唯一分子标识符 (UMI,具有标记单个 mRNA分子的随机核苷酸序列,用于量化唯一转录本和纠正扩增偏好性);(ii)唯一的细胞条形码(barcode);(iii)Illumina 接头;和(iv)T7 启动子。将扩增的 RNA 分子重新转换为 cDNA需要另一轮逆转录,以便构建测序文库。因此,得到的 cDNA 在 3′端的覆盖度上存在明显的偏差。此外,这种方法耗时耗力,使之与 PCR 相比,没有得到广泛的应用(Hashimshony 等人,2012)。
作为 scRNA-seq的一个独特步骤,所有来自单个细胞的转录本都会获得一个独特的barcode-在逆转录期间引入 cDNA 的短寡核苷酸序列,用于区分单个细胞的转录组。利用独特的barcode信息,这些转录本就可以被很容易地对应到各自归属的细胞中去。因此,在每个细胞都用唯一的barcode标记后,大量单细胞转录组才能被合并在一起,用于构建文库,然后在一次运行中完成后续测序。这种方法显著降低了成本并提高了测序通量。细胞barcode是一种非常有效的并行处理策略,2011首次用于 STRT-seq方法(Islam等,2011)。随后,2014, Islam 等人进一步创新,引入了 UMI 来识别细胞内的每个 cDNA 分子。与细胞barcode一样,UMI 也是一个由 6–8 个碱基对组成的随机寡核苷酸序列,可以在逆转录过程中整合到细胞内的每一个转录本中。大量的UMI序列可以让每个转录本都带上不同的标记,从而确保通过 PCR 扩增产生的所有重复分子都保留原始转录本的UMI。通过计算UMI,就可以对转录本的原始拷贝数进行精准的定量,从而消除 PCR 扩增的偏好性。
针对单细胞 RNA 测序的三个关键问题:(i)单细胞分离;(ii)最大限度地减少逆转录期间的RNA损失;以及 (iii)产生足够的DNA进行测序,研究人员近年来进行了许多技术方面的探索,导致了各种 scRNA-seq方法的提出,专门用于单细胞转录组学的全面研究。
不同的 scRNA-seq技术都基于共同的基本原理,但它们在以下至少一个方面有所不同:(i)单细胞分离;(ii)逆转录;(iii)cDNA 扩增;(iv)转录本覆盖;(v)UMI;(vi)链特异性。每种方法都有其独特的优势和局限性。这些不同的 scRNA-seq技术的主要特征合并整理在表S1 中。因此,研究人员可以根据技术特点、优势、成本因素和通量需求灵活地选择合适的 scRNA-seq方法。
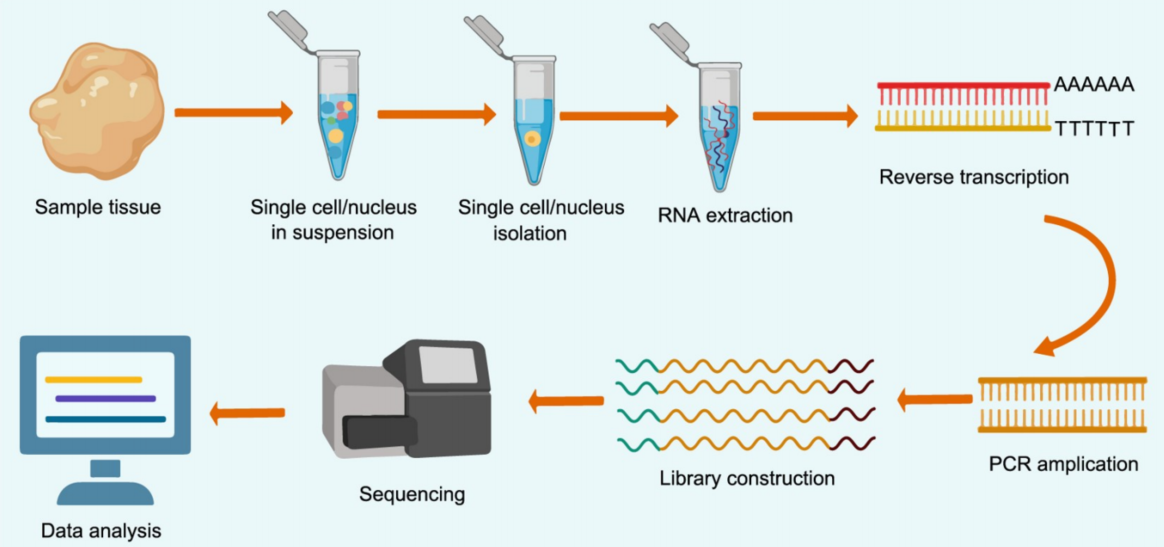
图3. 单细胞测序工作流程的主要步骤
当前可用的 scRNA-seq 技术
低通量 scRNA-seq 技术
如表S1 中所述,scRNA-seq已经开发出了60多种不同的方法。总体来说,这些方法可以分为两大类:低通量方法和高通量方法。低通量方法发展出的基本化学方法旨在提高测序的灵敏度和准确性,同时降低成本和技术噪音。其中,灵敏度是最为重要的关键特性,也是评估方法性能的基本指标。高通量方法的基本化学原理也是从几种经典低通量方法中发展而来的。我们将在下面详细讨论这些技术。
(1)Tang's protocol
Tang's protocol于2009年由Surani课题组开发,是一种具有开创性的scRNA-seq方法。在该技术中,单个小鼠胚泡是利用一台显微镜来手动进行选择的。随后解列细胞,用带有锚定序列(UP1)的 poly(T)引物将 mRNA 逆转录为 cDNA,并在末端转移酶的帮助下将 poly(A)尾添加到第一链末端的尾端。此后,使用带有另一个锚定序列(UP2)的第二条 poly(T)引物合成第二链 cDNA。在此之后,使用 UP1 和 UP2 引物通过PCR对cDNA进行高效扩增,构建文库,最后在SOLiD系统上进行测序。该方法可以生成几乎全长的转录本 cDNA 并检测约 13,000 个基因(Tang等,2009)。该技术的主要用途是帮助揭示新的转录本和可变剪接异构体。
尽管该技术代表了当时新兴的scRNA-seq领域的重大进步,但它也有很多局限性。首先,该方法只能识别具有poly(A)尾的 mRNA;它不能捕获没有poly(A)尾的 mRNA,例如组蛋白 mRNA、miRNA、环状 RNA(circRNA)和新生 RNA。这是因为该方法需要依赖于poly(T)引物通过poly(A)尾来捕获mRNA,进而起始逆转录反应。其次,酶反应效率低下导致测序灵敏度降低,从而导致低表达转录本的丢失。第三,这种方法不是链特异性的,不能区分正义和反义转录本。因此,它没有被广泛使用。
(2)STRT-seq
2011年,Islam 等人(2011)开发了一种单细胞标记逆转录测序 (single-cell tagged reverse transcription sequencing,STRT-seq)方法,这是一种基于Illumina 平台多重测序的scRNA-seq方法。该方法在逆转录过程中通过模板切换机制引入barcode和上游引物结合序列,促进 3′端链特异性扩增以及高通量96细胞多重测序。STRT-seq在基于阵列的测序策略基础上,支持处理多达 800 个单个细胞。该方法的一个关键优势是它可以在逆转录过程中为每个细胞添加独特的barcode序列,从而能够大规模检测各种混合细胞样本,例如高度异质性的肿瘤细胞样本。与Tang’s protocol相比,STRT-seq通过提前加入barcode的策略显著降低了成本和处理时间。然而,该方法中包含了多个PCR循环,可能会引入PCR偏差。该方法在生物医学领域有许多应用,包括探究肿瘤异质性、识别潜在的用于疾病的诊断和治疗的新型生物标志物或者药物靶点(Cui等,2021 ;Song等,2022a ;Tian等,2022)。(3)Smart-seq
2012, Ramsköld 等人开发了一种可靠、稳定的 scRNA-seq方法 Smart-seq,该方法最显著的进步是通过转录本前端的切换机制,在转录本全长 cDNA 合成中达到超过40%的效率。该技术的发表是 scRNA-seq研究领域的一个里程碑。Smart-seq的核心原理是利用 poly(T)引物和 SMART-TS 技术将多聚腺苷酸化(poly(A)+)RNA 转换为全长 cDNA。得到的cDNA分子经PCR扩增后,通过Nextera Tn5转座子技术构建Illumina测序文库,该方法显著提高了检测可变剪接外显子和低丰度表达转录本的能力。smart-seq方法在医学领域有着广泛的应用,如分析胆道闭锁小鼠模型肝脏中CD177+细胞的基因表达谱(Zhang等,2022d);分析骨骼肌干细胞、不同生态位的细胞和单根肌纤维的全基因组表达(Blackburn等,2019 ;Blackburn等,2021);研究急性皮肤红斑狼疮患者与正常对照组真皮CD4 + Trm细胞的差异(Zhao等,2022c)。
Smart-seq2被开发出来是为了克服 Smart-seq中存在的覆盖范围有限、cDNA扩增产量较低和灵敏度不够等问题(Picelli 等人,2013)。为了增加 cDNA 文库的产量和长度,Smart-seq2进行了几项改进,包括增强逆转录、TSO 和PCR重新扩增。与 Smart-seq相反,Smart-seq2是通过在 TSO 的 3 '端加入锁核酸 (LNA)鸟苷酸,使得cDNA 产量显著提高至大约两倍。这种改善与LNA-DNA碱基对的热稳定性增强有关。此外,该技术通过添加甲基供体甜菜碱以及更高浓度的 MgCl2,使得cDNA 产量大幅增加。另一方面,不直接将脱氧核糖核苷三磷酸 (dNTP)加入RT主混合物中,而是在RNA 变性之前才加入反应体系,这样可以增加预扩增cDNA的平均长度。上述改进可能是由RNA 与oligo-dT 引物杂交稳定性提高所导致的。KAPA HiFi Hot Start DNA 聚合酶的应用改善了cDNA的生成并增加了cDNA的长度。Smart-seq2 转录组文库在检测强度、覆盖率、偏差和准确性方面均优于 Smart-seq。Smart-seq2 转录组文库的建立可以使用成本更低的现成试剂,使得深入分析每个转录本的全外显子和检测不同的剪接变体成为可能。该方法还有助于彻底分析单核苷酸多态性 (SNP)和突变。然而,该方法也存在局限性,例如缺乏链特异性和无法检测 poly(A)− RNA(Picelli等,2014)。此外,使用微量移液器的细胞分离过程较为耗时且通量低。
(3)Smart-seq3
通过优化逆转录和模板转换的条件,提高了灵敏度(HagemannJensen等,2020)。优化的参数包括使用Maxima H-minus逆转录酶、将逆转录盐从KCl转换为NaCl或CsCl、在体系中存在5% 聚乙二醇 (PEG)的条件下进行逆转录,以及加入GTP或dCTP来增强和稳定模板转换反应等。Smart-seq3 的一个显着特点是它将全长转录组覆盖与 5′UMI RNA计数策略相结合,从而无需牺牲整体覆盖率即可提高转录本计数的精度。这种方法构建了一个TSO引物,包含Tn5基序的一部分、11 个碱基对的标签序列、8 个碱基对的 UMI 序列和三个核糖鸟苷。测序后,利用上述11 个碱基对的标签就可以明确区分 5′UMI 标记的reads和内部reads,在单次测序反应中就可以收集5′UMI标记的reads和不带 UMI 的整个转录本的内部reads。这种方法允许利用UMIreads对原始转录本进行准确量化,从而校正非线性PCR扩增偏差,通过使用内部reads就可以重建全长转录本。当前的很多测序文库构建方法结合了细胞barcode和 UMI 标签策略。然而,由于这些标签只能引入到cDNA的末端,因此得到的用于测序的cDNA序列仅限于转录本的一端,导致转录本中间的重要序列信息丢失。因此,基于标签的方法主要用于基因表达的量化,不适用于异构体识别或剪接的研究。尽管 Smart-seq和 Smart-seq2 能够捕获全长转录本,但它们无法利用barcode或 UMI 来对转录本进行标记,这就使得它们与高通量并行的单细胞测序不兼容。此外,如果没有 UMI 标签,这些方法也就无法解决PCR带来的扩增偏好性问题。然而,Smart-seq3 通过使用一种特殊的TSO引物克服了全长转录本覆盖和UMI不兼容的限制。
Smart-seq-total被设计出来是为了针对之前的 Smart-seq技术只能捕获 poly(A)+ RNA 分子的限制 ( Isakova等,2021 )。这种方法的主要进步在于利用大肠杆菌 poly(A)聚合酶在 RNA 分子的 3′端添加腺嘌呤尾巴。因此,所有 poly(A)+ RNA在进行逆转录时使用的poly(T)引物都包含UMI和TSO。这种修改使 Smart-seq-total 能够同时捕获多种形式的RNA,包括单个细胞内的蛋白质编码RNA、长非编码RNA、microRNA 和其他非编码RNA转录本。这种方法有助于探索细胞内编码和非编码转录本之间的调控联系,从而深入了解复杂的调控景观。但值得注意的是,Smart-seq-total确实也存在一些局限性。首先,它不能评估环状RNA。其次,它会导致转录本内源性多聚腺苷酸化状态的丧失。尽管存在这些缺点,Smart-seq-total 仍然表现出巨大的潜力,可以揭示控制细胞功能的非编码调控模式,并有助于确定细胞身份。
(4)CEL-seq
CEL-seq(Cell expression by linear amplification and sequencing)是把IVT 中的线性的链特异性用于扩增单细胞中的 RNA 的开创性方法 ( Hashimshony 等,2012 )。该过程的起始包括使用具有锚定 poly(T)、特定barcode、5′Illumina 测序接头和 T7 启动子的引物合成cDNA的第一链。随后生成第二条链,产生含有 T7 启动子的双链cDNA。来自多个细胞的混合cDNA样本由T7启动子起始IVT,从而实现 cDNA 的线性扩增,得到扩增后的RNA随后转化为cDNA 进行测序。通过线性扩增,CEL-seq最大限度地减少了扩增偏差,与Smart-seq等全长 cDNA 覆盖技术相比,可提供更灵敏、更可重复的结果。然而,CEL-seq也有局限性,例如无法检测 miRNA 和其他 poly(A)−转录本,并且由于其强烈的 3′偏差而难以区分不同的可变剪接形式。
CEL-seq2 是 CEL-seq的改进版本,它提高了灵敏度、降低了成本并减少了动手时间(Hashimshony 等人,2016)。为了减轻mRNA分子计数的偏差,CEL-seq2 在barcode上游引入了 5 个碱基对的 UMI。使用 SuperScript II 双链 cDNA 合成试剂盒并改变CEL-seq引物长度,逆转录反应效率显著提高。CEL-seq2优于原始 CEL-seq方法,每个细胞可检测到两倍多的转录本,检测到的基因数增加30%。但值得注意的是,尽管 CEL-seq2 具有明显的3′偏向性,但是大多数剪接事件的信息它都不能提供。尽管如此,其更高的灵敏度和单个转录本计数能力使其再转录组学中的各种应用具有更多的优势。
(5)SUPeR-seq
SUPeR-seq(single-cell universal poly(A)-independent RNA sequencing)使用具有固定锚序列的随机引物代替 cDNA 合成中常用的 oligo-dT 引物,这使得检测单个细胞内的poly(A)+ 和 poly(A)− RNA成为了可能(Fan等,2015b)。该过程涉及利用具有固定锚定序列的随机引物(AnchorX-T15N6)将总 RNA 逆转录为cDNA第一条链。在合成初始的cDNA链后,用ExoSAP-IT 消化多余的引物,防止形成引物二聚体复合物。使用末端脱氧核苷酸转移酶和含有1% ddATP 的 dATP将 poly(A)尾巴附加到初始 cDNA 链的 3′端,随后,使用具有可变锚定序列的 poly(T)引物(AnchorY-T24)生成cDNA的第二条链。上述两条链使用 AnchorY-T24 和 AnchorX-T15 引物通过 PCR 扩增并进行测序。SUPeR-seq已被用于研究哺乳动物早期胚胎发育过程中 circRNA 的调控机制。然而,由于缺乏UMI和细胞barcode,它给高通量测序和分子计数带来了挑战。
(6)MATQ-seq
2017年,Sheng 等人(2017)介绍了基于多重退火和加dC尾的定量单细胞 RNA 测序(MATQ-seq)方法。有别于SUPeR-seq,该技术结合barcode和UMI对polyA+和polyA − RNA进行测序。该过程包括使用为多重退火和基于环状的扩增循环(MALBAC)设计的引物将总RNA转化cDNA第一条链。这些引物主要包含G、A和T碱基,以及MALBAC-dT引物。逆转录后,cDNA一链进行dC加尾,然后使用G富集的MALBAC引物合成cDNA二链。UMI在第二链合成过程中添加。与 Smart-seq2和SUPeR-seq不同的是,MATQ-seq利用UMI 显著减少了HEK293T 转录本中的前导端或尾端偏差。此外,MATQ-seq在捕获 polyA − RNA 方面表现出比 Smart-seq2 和 SUPeR-seq更高的灵敏度,捕获效率为 89.2%±13.2%。这一改进提高了检测低丰度基因的效率。MATQ-seq的高准确度和灵敏度可以检测到同一类细胞中各个细胞之间基因表达的细微差异。然而,与 SUPeR-seq类似,MATQ依靠口吸管进行单细胞分离的方法十分耗时,限制了 MATQ-seq的通量。
(7)FLASH-seq
FLASH-seq是一种快速且透彻的全长 scRNA 测序方法,由Hahaut 等人(2022)开发。为了提高 Smart-seq2方法的效率,该技术进行了几个关键的修改:(i)将逆转录和 cDNA 预扩增结合起来,简化了流程;(ii)用更高效的逆转录酶 Superscript IV取代了Superscript II,缩短了逆转录反应时间;(iii)增加了dCTP的量,有利于Superscript IV 的 C 尾活性并增强模板转换反应;(iv)将模板转换寡核苷酸中的 3′-末端锁核酸鸟嘌呤替换为核糖鸟苷;(v)将反应体积减少到 5 μL ( Hahaut等,2022 )。这些改动共同导致时间和成本大幅降低。FLASH-seq可以在大约 4.5 小时内完成,比 Smartseq2 等其他方法快2–3.5 小时。与 Smart-seq3 相比,它的单个细胞成本低于其他商业化和非商业化的方法,不到 1 美元。此外,FLASH-seq可以检测到大量的 SNP。该方法适合于寻求经济实惠、自动化友好、稳定且高效的单细胞转录分析方法的研究人员。
高通量scRNA-seq技术
(1) 开发高通量 scRNA-seq的策略
ScRNA-seq的早期阶段使用显微操作器或 LCM 分离单个细胞,进行单独的转录组扩增和文库构建。然而,这些方法有局限性,因为它们在一次实验中只能分析几个细胞。细胞特异性barcode的引入彻底改变了该领域,允许将数千个单细胞转录组混合在一起,在单次运行中进行文库构建和测序,从而实现高通量并行测序。该方向上的一个显著进步是 MARS-seq方法的开发,该方法将 FACS 与自动液体处理相结合,在一次实验中成功测序数千个细胞(Jaitin 等人,2014)。该方法采用三种不同水平上的标识用于标记细胞、培养板和mRNA,便于混合所有材料进行后续自动化处理。与 MARS-seq类似,STRT-Seq-2i 旨在通过实施专门的 FACS 和barcode流程来提高测序通量(Hochgerner 等人,2017)。该方法使用定制的铝板,该铝板上有 9,600 个孔,排列成 96 个子阵列,每个子阵列有 100 个孔,一次运行即可同时对 9,600 个细胞进行测序。然而,尽管这些基于培养板的方法提高了通量,但可分析的细胞数量仍然有限。微流控技术的引入从根本上解决了高通量单细胞操作带来的挑战。2012,Fluidigm C1 系统成为首个商用自动化微流控平台,可同时对 96 个单细胞进行自动细胞分离、细胞裂解、cDNA 合成、扩增和文库制备。但该系统的处理能力有限,无法满足高通量并行测序的需求。2015年,基于液滴的微流控 scRNA-seq技术的出现带来了革命性的突破。以Klein 等人和Macosko 等人开发的方法为例,这类方法能够同时处理数千个细胞(Klein等,2015 ; Macosko等,2015)。这项重大进步使单细胞基因组学领域真正大规模并行测序成为可能。随后,其他高通量并行测序策略也被开发出来,包括 sci-RNA-seq(Cao等,2017)和 SPLiT-seq(Rosenberg等,2018)。这些方法采用组合式索引方法来标记细胞,而无需对单个细胞进行物理分隔。这些技术具有很高的细胞标记效率,操作简单,并可大幅降低测序成本。我们将在下文详细讨论这些创新技术。
(2)基于微孔板的高通量 scRNA-seq方法
基于微孔板的高通量 scRNA-seq方法包括通过 FACS 将细胞分选到微孔板上,并标记细胞转录本以进行后续高通量测序。这些方法的优势在于可以处理任意数量的单个细胞,而不会显著影响每个细胞的成本。另一方面,其他高通量方法,例如基于液滴的技术,如 Drop-seq(Macosko等,2015)、InDrop(Klein等,2015)和 10x Chromium(Zheng等,2017),在同时分析大量细胞时能够分摊成本,使成本的效益更高。基于孔板的方法的测序通量受所用微孔板数量的限制。接下来将详细讨论这些技术。
1)Quartz-seq。为了揭示非遗传细胞异质性的生物学功能和根本原因,Sasagawa 等人开发了简单但定量水平很高的Quartz-seq技术。Quartz-seq通过解决三个关键方面提高了全转录本扩增的简单性和定量性能。首先,为了克服以往基于 poly(A)尾反应的全转录本扩增技术中副产物过多的问题,Quartz-seq结合了外切酶 I 处理、调控 poly(A)尾以及优化的阻抑PCR。这种策略方法完全消除了副产物的合成,简化了后续的 scRNA-seq分析。其次,该技术采用了针对单管反应优化的强效 DNA 聚合酶 (MightyAmp DNA Polymerase)。这种 PCR 酶的选择提高了 cDNA 产量,提高了全转录本扩增复制的可重复性,并减少了所需的 PCR 循环次数。最后,Quartz-seq通过调节退火温度来优化 RT 和第二链合成的效率。值得注意的是,该方法的所有步骤都合并到一个 PCR 管中,无需纯化,每个单细胞仅涉及六个反应步骤。这种精简大大简化了 Quartzseq方法,有助于其高通量实施。除了简单之外,Quartz-seq还表现出很高的定量准确性、可重复性和灵敏度。因此,它可以辨别各种类型的非遗传细胞异质性,并区分不同细胞类型和同一细胞类型内的细胞周期阶段。
Quartz-seq2 ( Sasagawa等,2018 )是一种创新的高通量 scRNA-seq方法,是作为 Quartz-seq的扩展而开发的。该方法包括使用 FACS 将单个细胞分选到 384 孔板中,然后使用结合了 oligo-dT 序列、细胞barcode序列和 UMI 序列的引物进行 RT。通过应用多个分子生物学阶段的进步,包括对 poly(A)标记的重大升级,Quartz-seq2 实现了出色的 UMI 转换效率。值得注意的是,Quartz-seq2采用了基于T55缓冲液和递增温度条件组合的poly(A)标记策略,使cDNA的数量增加了约3.6倍。
Quartz-seq2的UMI转换效率非常高,从32%到35%不等,超过了CEL-seq2、SCRB-seq和MARS-seq(7%-22%)等其他方法。由于效率的提高, Quartz -seq2可以从更少的序列读取中以更低的成本识别出更多的转录本。与MARSseq类似,Quartz-seq2使用FACS进行细胞分选,这一过程需要熟练的工人。尽管有这个要求,但该技术更高的UMI转换效率和成本效益使其成为scRNA-seq应用的一种有价值方法。
2)MARS-seq。MARS-seq是一种自动化、高度并行的RNA单细胞测序技术,开发于 2014(Jaitin等,2014),通过在 oligo-dT 引物中引入UMI实现对唯一RNA分子的计数,从而彻底改变了这一领域。MARS-seq流程使用 FACS 将单细胞分选到384 孔板中。逆转录和文库构建过程遵循 CEL-seq流程,确保方法具有系统性和可重复性。MARS-seq的关键创新之一在于其自动化,该方法的每一步都由液体处理机器人执行。这种自动化提高了该技术的可重复性,并显著提高了通量。MARS-seq的高通量和自动化特性使其适用于正常和疾病状态下的各种组织和器官。通过描述组织的细胞类型和细胞状态组成,MARS-seq有助于全面了解这些生物实体,并将这些信息与详细的全基因组转录谱联系起来。
2019,研究团队在 MARS-seq方法的基础上,开发了用于索引排序和大规模并行单细胞 RNA 测序的集成流程 (MARS-seq2.0)(Keren-Shaul等,2019)。MARS-seq2.0 能够在一次运行中高效测序 8,000–10,000 个细胞,显著提高了通量。值得注意的是,该方法将逆转录反应的体积减少了八倍,从 4 μ L 减少到 500 nL。由于体积减少,制备单细胞文库的成本将下降六倍。MARS-seq2.0 是一种基于 3′的 scRNA-seq技术,这限制了它在确定基因 5′端的可变剪接异构体或特定序列方面的应用。这是需要记住的关键一点。尽管存在这一限制,MARS-seq2.0 中索引式 FACS 分类与 scRNAseq的整合已被证明有利于识别稀有亚群和处理人类临床样本中的稀有细胞。此外,MARS-seq2.0 提供了一个灵活的平台,通过将无偏转录图谱与大量荧光标记相结合,能够同时获得同一单细胞的多层信息,涵盖遗传学、信号传导、表观遗传学和空间定位。这种多方面的方法有助于更深入地从分子层面理解生理过程和疾病。
3)SCRB-seq。为了降低探究异质性细胞群中基因表达变异的主要模式的成本, Soumillon 等人基于 Smart-seq方案开发了SCRB-seq(single-cell RNA barcoding and sequencing),使用最少的试剂和最少的单个细胞测序读数来分析大量细胞中的mRNA。SCRB-seq利用FACS将单个细胞分选到 384 孔板中。Poly(A)+ mRNA 通过 RT 转化为 cDNA,逆转录使用模板转换逆转录酶和由barcode、UMI 和 poly(T)引物组成的逆转录引物。此后,链信息被保留下来,来自多个细胞的被修饰过的cDNA混合在一起进行扩增,用改进的基于转座子的片段化技术进行测序,该技术富集 3′端。SCRB-seq技术能够测序大约 12,000 个单细胞,提供深度和全长转录组覆盖测序。此外,它所需的酶反应、纯化和液体转移步骤比 MARSseq方法少大约两倍 ( Jaitin等,2014 )。与 Smartseq相比,SCRB-seq在逆转录期间添加了独特的细胞barcode,从而更容易识别来自同一细胞的读取并提高测序通量。尽管如此,对大量单细胞进行测序仍然面临挑战。
4)STRT-seq-2i。STRT -seq-2i 是一种双索引 5′单细胞和细胞核 RNA 测序方法,旨在显着提高通量 ( Hochgerner等,2017 )。它使用专门设计的 9,600 微孔板,有助于提高效率。微孔阵列可以对单细胞孔进行成像验证,减少单孔中双联体的出现。除了保留了一些优点,例如可显示转录起始位点的 5′端reads、添加 UMI 进行绝对定量,以及使用单端测序而非双端测序以最大限度提高成本效率之外,STRT-seq-2i 仍然与之前描述的 STRT-seq方法兼容。该技术已证明其可适应各种实验环境,因为它可用于检查新鲜单个小鼠皮质细胞和冷冻的死后人类皮质细胞核的转录谱。
(3)基于微流体的高通量 scRNA-seq方法
基于液滴和微孔的平台是高通量 scRNA-seq的主要技术能够在一次实验中分析大约 10,000 个单细胞的转录组。这两种方法基本上都涉及将单个细胞分离到许多纳升大小的容器(例如微孔或油包水液滴)中,其中包含逆转录所需的化学物质。与非微流体方法和带有阀门的微流体方法相比,将细胞barcode集成到这些微流体平台中的策略可显著提高通量,同时降低成本。这一进步对于需要对大量细胞进行全面转录组分析的生物医学研究应用尤其有利。
1)基于液滴的 scRNA-seq技术。(i)InDrop。inDrop 的基本技术(Klein 等人,2015)是将单个细胞包裹在含有裂解缓冲液、逆转录试剂和barcode水凝胶微球 (BHM)的液滴中。每个 BHM 包含 ~109个可以光解的标签引物(147,456 个不同的标签)。该技术中采用的微流体装置包含用于载体油、细胞、裂解/逆转录试剂和 BHM 的入口,以及用于液滴收集的出口。每个 BHM 都通过光释放键共价连接到标签引物,这些引物具有 T7 RNA 聚合酶启动子、Illumina 测序接头、独特细胞barcode、UMI 和 poly(T)尾。样本中的所有 BHM 共享相同的细胞barcode以区分样本,但拥有不同的 UMI 以进行精确的转录计数。封装后,紫外线 (UV)暴露使引物释放并结合mRNA ,在 cDNA 合成过程中进行barcode编码,同时使反应仍限制在同一液滴中。随后,按照 CEL-seq方案(Hashimshony 等,2012),在液滴破裂后汇集所有细胞的 cDNA 以进行文库构建和测序。与传统方法不同,InDrop固有的可扩展性不受反应室数量的限制。此外,通过在液滴内进行裂解和逆转录简化了操作过程。然而,inDrop 的一个显著缺点是其相对较低的 mRNA 捕获效率(~7%),这使得转录本丰度低于 20–50 个转录本的基因在单细胞中难以可靠地检测到。
(ii)Drop-seq。与inDrop 类似,Drop-seq旨在对数千个单细胞中的 mRNA 表达进行高通量分析。它通过将每个细胞与带有唯一标记的珠子共同封装在纳升级的油包水液滴中以同时处理来实现这一点(Macosko等,2015)。与使用标记水凝胶微球的 inDrop 不同,Drop-seq使用由不变的硬树脂制成的珠子。这些树脂珠直接用标记引物合成,其中包括用于捕获 mRNA 的 poly(T)序列、细胞barcode、UMI 和用于扩增的通用PCR序列。每个珠子含有超过 108种不同的引物。细胞在液滴中裂解后,释放的 mRNA 与伴随珠子上引物的poly(T)尾巴杂交,生成固定在微粒上的单细胞转录组 (STAMP)。液滴破裂后,数千个 STAMP 汇集在一起,在一次反应中进行逆转录、PCR 扩增和测序。与 inDrop (147,456)相比,Drop-seq方法拥有更多独特的barcode (16,777,216),从而能够实现经济高效且快速的高通量分析。
(iii)10x Genomics。10x Genomics 系统 ( Zheng等,2017 )是一个完全商业化的平台,与 inDrop 和 Drop-seq有相似之处。反应体系中的凝胶珠 (GEM)是该方法的基本组成部分。创建 GEM需要使用 8 通道微流体装置,该芯片在6分钟内可以生成 100,000 个 GEM,每个 GEM 可容纳数千个细胞。GEM 中的每个凝胶珠都含有标签寡核苷酸,包括 Illumina 接头、10x barcode、UMI 和 poly(T)尾巴,使得poly(A)+ RNA能够逆转录。细胞与凝胶珠共封装成液滴后,细胞立即裂解,释放出mRNA。随后,凝胶珠溶解并释放带标签的寡核苷酸,从而实现poly(A)+RNA的RT。RT反应在每个液滴内进行,产生的cDNA分子具有每个GEM共享的barcode、唯一的UMI,并在3′端以TSO结尾。去除油相后,带标签的cDNA混合起来,遵循Smart-seq流程进行PCR扩增。
InDrop、Drop-seq和Chromium是三个类似的平台,它们采用液滴微流控方法分离单细胞以进行高通量测序。这三种方法每天都可以快速处理数万个细胞。这三种技术在以下四个方面有所不同:
首先,inDrop和10x Genomics使用的是水凝胶微球,而Drop-seq中使用的珠子是硬树脂。inDrop 和 10x Genomics 使用的水凝胶珠子较柔软,封装在微流控通道中时可以同步产生超泊松分布。对于单个细胞和珠子在同一个液滴中的封装,双泊松分布控制着微小硬珠子的封装。因此,inDrop 和 10x Genomics 可以获得比 Drop-seq更高得多的细胞捕获效率。据报道,10x Genomics 的细胞捕获率约为 50%(Zheng等,2017)。
其次,inDrop 和 10x Genomics 使用水凝胶珠子,可以将引物固定在珠子内部,而 Drop-seq的引物只能固定在更小的硬珠表面。封装后,inDrop 利用紫外线诱导的裂解来释放引物。10x Genomics通过直接溶解珠子将引物全部释放到溶液中,提高了mRNA的捕获效率,而Drop-seq的引物不能从珠子中释放出来,mRNA分子会与珠子上的poly(T)尾杂交形成STAMP进行RT,这是Drop-seq相较于inDrop和10x Genomics的一个缺点。再次,Drop-seq在mRNA与引物杂交后立即分裂液滴,将所有STAMP混合在一起进行RT,而inDrop和10x Genomics方法将RT试剂共封装在液滴中,每个液滴内独立进行RT反应,有利于提高cDNA转化的特异性,提高相对产量,减少试剂消耗(Streets等,2014)。
最后,三个平台采用了不同的建库策略。Drop-seq和 10x Genomics 使用类似于众所周知的 Smart-seq化学的模板切换程序,而 inDrop 使用 CEL-seq方法。因此,它们分别继承了 Smart-seq和 CEL-seq的优点和缺点。
(iv)MULTI-seq。为了在逆转录期间传递细胞特异性barcode,之前讨论的所有基于液滴的技术通常都使用共封装策略,这需要同时封装细胞和barcode珠子。MULTI-seq方法(使用脂质标记进行单细胞和单核 RNA的样本多重测序)最近由McGinnis 等人 (2019b)引入,作为一种以独特方式使用细胞barcode的替代策略。在这项技术中,DNA barcode通过与“锚”脂质修饰寡核苷酸 (LMO)杂交而被标记到单细胞的质膜上。“锚定” LMO 的疏水性 5 '二十六酸酰胺与膜结合;与带有 3 '棕榈酸酰胺的“共锚定” LMO 杂交可增强复合物的疏水性,延长膜保留时间。3'poly(A)捕获序列、8 bp 样本barcode和 5'PCR 手柄组成 LMO。LMO 携带的每个单细胞或细胞核与 mRNA 捕获珠共同封装到由 10x Genomics 系统生成的乳液液滴中。样本解复用是通过在液滴内细胞裂解时释放内源性 mRNA 和 LMO 实现的,它们在逆转录期间都与 mRNA 捕获珠杂交并附着到通用细胞barcode上。内源性 mRNA 和 LMO 在扩增后通过大小选择分离,从而实现以用户定义的比例进行汇集测序。使用此方法可以对任何具有可及质膜的物种的任何细胞类型或细胞核进行barcode编码。此外,这种方法涉及的样品处理最少,从而保留了细胞活力和内源基因表达模式。
2)基于微孔的 scRNA 测序方法。(i)CytoSeq。Cytoseq是一种高度可扩展的 scRNA 测序方法,可同时分析几千个细胞,并可轻松扩展到 10,000 或 100,000 个细胞,每个细胞可检测大约 100 个基因(Fan等,2015a)。该方法采用递归泊松策略来调整悬浮液中的细胞数量,促进细胞在重力作用下高通量沉降在 100,000 个微孔中的 1/10 中。由于反应中使用的体积为纳升,因此文库制备成本极低。此外,Cytoseq的优势在于它不受特定细胞大小和形状的限制,可以研究大量异质细胞群的表达谱。这种灵活性允许在大量背景群体中检测稀有细胞类型。
(ii)Seq-Well。Seq-Well 是一种便携、经济高效、用户友好且高效的 scRNA-seq方法,专为低输入样本而设计(Gierahn等,2017)。该方法利用皮孔阵列,其中装有barcode mRNA 捕获珠和细胞,每个孔可容纳一个细胞和近一个珠子。细胞在重力作用下沉入孔中后,半透膜会密封阵列,为每个孔创建独特的环境,允许缓冲液交换但防止大分子迁移。随后,细胞被裂解,并进行扩增和测序过程。Seq-Well 板有大约 86,000 个亚纳升孔,可以同时分析来自不同来源的数千个细胞中的转录组。该方法特别适合于低样本输入,例如组织夹、细针抽吸物以及难以研究的细胞,如肝细胞和粒细胞(Kumar,2021)。值得注意的是,Seq-Well的实现只需要一个皮孔阵列、一个移液器、一个聚碳酸酯膜、一个烤箱或热源、一个夹具和一个管旋转器即可生成稳定的 cDNA 产物。这种简单性使其能够适应资源有限的环境,例如诊所和偏远地区(Aicher 等人,2019)。
(iii)ICELL8。ICELL8 是一种基于微孔的微流体系统,通过结合一个包含 5,184 个纳米孔的微芯片来提高吞吐量,从而可以捕获和处理大约 1,300 个单细胞(Goldstein 等人,2017)。每个纳米孔均含有预印的寡核苷酸,包括寡核苷酸 (dT 30 )引物、孔特异性barcode序列和 UMI。该过程包括使用多样品纳米分配器 (MSND)将单细胞悬浮液分配到微芯片纳米孔中,然后通过冻融循环裂解细胞。细胞裂解后,进行逆转录以合成 cDNA,采用 SCRB-seq方法。最终,将来自数百个细胞的 cDNA 汇集到一个试管中用于构建文库。该方法有几个优点:i)使用 MSND 仪器将细胞准确地分配到纳米孔中,消除了与手动移液相关的误差;ii)结合成像软件来识别含有活单细胞的纳米孔,确保只有含有单细胞的孔才会被处理到测序文库中,从而产生较低的细胞多重率 (<3%); iii)使用多样本纳米分配器在一个阵列上加载多达八种实验条件的能力,使得在一次实验中,一块芯片上 5,184 个纳米孔可同时处理 800–1,400 个细胞。
为了提高 ICELL8 的捕获率,并能够同时处理超过 5,000 个细胞用于测序文库, Shomroni 等人 (2022)介绍了一种名为 CellenONE-ICELL8 的新型 scRNA-seq方法。该方法将 ICELL8 处理仪器与 CellenONE 分离和分选系统相结合。CellenONE 依靠基于图像的单细胞分离,能够在随后的样品处理和测序之前,根据细胞形态、大小或荧光标记等参数选择高度纯化的单个细胞。与单独的 ICELL8 系统相比,CellenONEICELL8 中两种仪器的集成显著提高了细胞捕获效率,捕获的细胞数量从典型的 1,200 到 1,400 个细胞增加到 3,300 多个细胞。此外,该方法利用SMART-seq技术,可以检测非编码RNA,特别是较长的基因间非编码RNA,以及内含子和基因间序列。
(4)基于组合索引的高通量scRNA-seq技术
1)Sci-RNA-seq。Cao等(2017)开发了第一个用于高通量scRNA-seq的组合索引方法,称为sci-RNA-seq。这种创新方法有助于分析大量单细胞或细胞核的转录组,通过使用双UMIbarcode提供3′覆盖和高深度测序。sci-RNA-seq的可扩展性允许在单次实验中生成约4×104个单个细胞转录组,索引规模高达576×960。这种可扩展性使得通过加入额外轮次的索引或/和使用更多带barcode的逆转录和 PCR 引物能够以亚线性成本扩展处理更多细胞。但sci-RNA-seq并非没有缺点,这些缺点包括繁琐的实验程序、高通量转座反应的高成本以及 FACS 分选带来的明显细胞损失。
2)SPLiT-seq。SPLiT- seq代表了另一种专为 scRNA-seq分析而设计的创新组合索引方法,可以检查保存在 1.33% 甲醛中的固定组织(Rosenberg等,2018)。与 sci-RNA-seq不同,SPLiT-seq通过连接将第二轮和第三轮barcode插入 cDNA 中。这种方法提供了更简单、更温和、更具成本效益的工作流程。SPLiT-seq的第一轮barcode可以充当样本标识符,从而实现多重并行的样本处理。SPLiT-seq特别适合分析临床相关组织样本中产生的固定、难以完全解离的细胞或细胞核。然而,应该考虑一些局限性,包括醛基细胞固定可能导致 mRNA 发生化学修饰、由于复杂的交联细胞内环境导致固定细胞内逆转录和连接反应效率不佳以及固定过程中 RNA 质量可能下降,从而导致检测到的基因水平降低。
单细胞核RNA测序
ScRNA-seq是探索复杂组织中细胞类型、功能过程和动态状态的有力工具。然而,在处理无法轻易分离的存档样本或组织时,其应用受到限制,从而阻碍了对新细胞类型或与免疫和疾病相关的关键信息的探索。为了解决这一困难,科学家们采用了单细胞核RNA测序 (snRNA-seq)技术,其中 RNA 测序实验使用细胞核而不是整个细胞作为代理进行。已经开发了几种 snRNA-seq方法来分析从冷冻、轻度固定或新鲜组织中获得的单细胞核RNA,包括 DroNc-Seq(Habib 等人,2017 )、Div-seq(Habib 等人,2016 )、snDrop-seq(Lake 等人,2019)。这些方法将转录组分析的适用性扩展到更广泛的样本类型和样本处理方式。
由于 snRNA-seq与传统 scRNA-seq方法检测到的基因之间存在高度相关性,因此 snRNA-seq技术在各个研究领域的使用已被证明很有价值(Fischer and Ayers,2021 )。这些技术可用于多种样本类型,例如新鲜组织,如脑 ( Affinati 等人,2021)、心脏 ( Nicin 等人,2021)、肾脏 ( Barwinska 等人,2021;Muto 等人,2021)、胰腺 ( Basile 等人,2021)、肌肉 ( Petrany 等人,2020)或脂肪组织 ( Sun 等人,2020b)、存档组织 ( Basile 等人,2021)、植物细胞 ( Conde 等人,2021)。尽管有这些优势,但与分离的细胞类型相比,分离的细胞核通常更具粘性。因此,应采取预防措施防止结块,否则会导致双峰率膨胀。此外,值得注意的是,某些基因转录本可能在 snRNA-seq和 scRNA-seq数据集之间表现出富集差异。例如,长链非编码 RNA(lncRNA)在 snRNA-seq数据集中富集,而位于细胞质中的线粒体转录本仅存在于 scRNA-seq数据集中(Fischer and Ayers,2021)。研究人员在选择单个细胞核进行测序之前,需要仔细评估这些亚定位基因对他们的特定研究项目是否至关重要。
参考文献:
Sun F, Li H, Sun D, et al. Single-cell omics: experimental workflow, data analyses and applications. Sci China LifSun F, Li H, Sun D, et al. Single-cell omics: experimental workflow, data analyses and applications. Sci China Life Sci. 2025;68(1):5-102. doi:10.1007/s11427-023-2561-0e Sci. 2025;68(1):5-102. doi:10.1007/s11427-023-2561-0